| Description: |
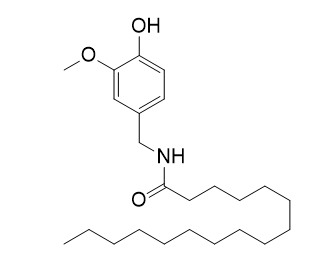
Palvanil is a TRPV1 channel activator and has antihyperalgesic activity.
Palvanil, may be promising agents in the therapy of IBS since it modulates intestinal motility and reduces visceral pain.
Palvanil inhibits inflammatory and neuropathic pain with little effects on bronchopulmonary function and body temperature. |
| In vitro: |
| Naunyn Schmiedebergs Arch Pharmacol . 2020 Aug;393(8):1357-1364. | | Desensitization of transient receptor potential vanilloid type-1 (TRPV1) channel as promising therapy of irritable bowel syndrome: characterization of the action of palvanil in the mouse gastrointestinal tract[Pubmed: 32002574] | | TRPV1 are involved in the control of the gastrointestinal (GI) functions and pain sensation. Their activation induces pain but it is followed by desensitization, which in turn causes analgesia. The studies from the last two decades indicate that TRPV1 are involved in visceral hypersensitivity in the GI tract and pathogenesis of irritable bowel syndrome (IBS). Therefore, the aim of this study is to assess the action of fast desensitizing agonist of TRPV1, palvanil (N-palmitoyl-vanillamine), in the murine GI tract and on nociception to evaluate its potential application in the therapy of IBS. The effect of palvanil on smooth muscle contractility was evaluated using organ baths. The impact of palvanil on intestinal secretion was assessed in Ussing chambers. In vivo, the action of palvanil (0.1-1 mg/kg) was assessed in whole GI transit, fecal pellet output, and colonic bead expulsion tests. The antinociceptive potency of palvanil was tested in the mustard oil-induced pain test. Palvanil inhibited colonic contractions (evoked by electrical field stimulation, EFS) and decreased the ion transport in the colon stimulated with forskolin. It did not affect secretion in experiments with veratridine. In vivo, palvanil prolonged whole GI transit at all doses tested. At the lower dose tested, it accelerated colonic motility during first 60 min following injection. By contrast, at the dose of 1 mg/kg, colonic motility was inhibited. Palvanil induced antinociceptive action at all tested doses in mustard oil-induced pain test. TRPV1 fast-desensitizing compounds, i.e., palvanil, may be promising agents in the therapy of IBS since it modulates intestinal motility and reduces visceral pain. | | Br J Pharmacol . 2019 Jul;176(13):2264-2278. | | Analgesic transient receptor potential vanilloid-1-active compounds inhibit native and recombinant T-type calcium channels[Pubmed: 30927254] | | Background and purpose: T-type calcium (Cav 3) and transient receptor potential vanilloid-1 (TRPV1) channels play central roles in the control of excitability in the peripheral nervous system and are regarded as potential therapeutic pain targets. Modulators that either activate or inhibit TRPV1-mediated currents display analgesic properties in various pain models despite opposing effects on their connate target, TRPV1. We explored the effects of TRPV1-active compounds on Cav 3-mediated currents.
Experimental approach: Whole-cell patch clamp recordings were used to examine the effects of TRPV1-active compounds on rat dorsal root ganglion low voltage-activated calcium currents and recombinant Cav 3 isoforms in expression systems.
Key results: The classical TRPV1 agonist capsaicin as well as TRPV1 antagonists A-889425, BCTC, and capsazepine directly inhibited Cav 3 channels. These compounds altered the voltage-dependence of activation and inactivation of Cav 3 channels and delayed their recovery from inactivation, leading to a concomitant decrease in T-type current availability. The TRPV1 antagonist capsazepine potently inhibited Cav 3.1 and 3.2 channels (KD < 120 nM), as demonstrated by its slow off rate. In contrast, neither the TRPV1 agonists, Palvanil and resiniferatoxin, nor the TRPV1 antagonist AMG9810 modulated Cav 3-mediated currents.
Conclusions and implications: Analgesic TRPV1-active compounds inhibit Cav 3 currents in native and heterologous systems. Hence, their analgesic effects may not be exclusively attributed to their actions on TRPV1, which has important implications in the current understanding of nociceptive pathways. Importantly, our results highlight the need for attention in the experimental design used to address the analgesic properties of Cav 3 channel inhibitors. | | Physiol Behav . 2018 Oct 15;195:158-166. | | Capsaicin produces antidepressant-like effects in the forced swimming test and enhances the response of a sub-effective dose of amitriptyline in rats[Pubmed: 30138635] | | Transient receptor potential vanilloid 1 (TRPV1) channels have been implicated in depression and anxiety. The aim of this study was to evaluate the antidepressant-like properties of the TRPV1 agonist capsaicin using the forced swimming test (FST) in rats. Capsaicin (0.001-0.25 mg/kg, i.p.) produced a reduction of immobility in the FST. A maximally effective dose of the tricyclic antidepressant amitriptyline (12 mg/kg) reduced immobility as well. Notably, doses of capsaicin (1 pg/kg, 1 ng/kg, and 0.001 mg/kg) that were ineffective when applied alone produced a significant decrease in immobility when combined with a subthreshold dose of amitriptyline (5 mg/kg). Rats treated with capsaicin (0.01 mg/kg) + amitriptyline (5 mg/kg) displayed less immobility than those treated with a maximally effective dose of amitriptyline. The non-pungent TRPV1 channel agonist palvanil (0.05-0.1 mg/kg, i.p.) also decreased immobility in the FST. Capsaicin (0.05 mg/kg) did not affect general locomotion in the open field test, nor performance in the elevated plus maze, or skeletal muscle contraction strength measured in vitro after the FST (at 0.25 mg/kg). Altogether, our results imply that low doses of capsaicin produce antidepressant-like effects, and enhance the effect of a subthreshold dose of amitriptyline in the FST. | | Br J Pharmacol . 2019 Jul;176(13):2264-2278. | | Analgesic transient receptor potential vanilloid-1-active compounds inhibit native and recombinant T-type calcium channels[Pubmed: 30927254] | | Background and purpose: T-type calcium (Cav 3) and transient receptor potential vanilloid-1 (TRPV1) channels play central roles in the control of excitability in the peripheral nervous system and are regarded as potential therapeutic pain targets. Modulators that either activate or inhibit TRPV1-mediated currents display analgesic properties in various pain models despite opposing effects on their connate target, TRPV1. We explored the effects of TRPV1-active compounds on Cav 3-mediated currents.
Experimental approach: Whole-cell patch clamp recordings were used to examine the effects of TRPV1-active compounds on rat dorsal root ganglion low voltage-activated calcium currents and recombinant Cav 3 isoforms in expression systems.
Key results: The classical TRPV1 agonist capsaicin as well as TRPV1 antagonists A-889425, BCTC, and capsazepine directly inhibited Cav 3 channels. These compounds altered the voltage-dependence of activation and inactivation of Cav 3 channels and delayed their recovery from inactivation, leading to a concomitant decrease in T-type current availability. The TRPV1 antagonist capsazepine potently inhibited Cav 3.1 and 3.2 channels (KD < 120 nM), as demonstrated by its slow off rate. In contrast, neither the TRPV1 agonists, Palvanil and resiniferatoxin, nor the TRPV1 antagonist AMG9810 modulated Cav 3-mediated currents.
Conclusions and implications: Analgesic TRPV1-active compounds inhibit Cav 3 currents in native and heterologous systems. Hence, their analgesic effects may not be exclusively attributed to their actions on TRPV1, which has important implications in the current understanding of nociceptive pathways. Importantly, our results highlight the need for attention in the experimental design used to address the analgesic properties of Cav 3 channel inhibitors. | | Pharmacol Res. 2011 Apr;63(4):294-299. | | N-palmitoyl-vanillamide (palvanil) is a non-pungent analogue of capsaicin with stronger desensitizing capability against the TRPV1 receptor and anti-hyperalgesic activity[Pubmed: 21215315] | | N-acyl-vanillamide (NAVAM) analogues of the natural pungent principle of capsicum, capsaicin, were developed several years ago as potential non-pungent analgesic compounds. N-oleoyl-vanillamide (olvanil) and N-arachidonoy-vanillamide (arvanil), in particular, were described in several publications and patents to behave as potent anti-hyperalgesic compounds in experimental models of chronic and inflammatory pain, and to activate both "capsaicin receptors", i.e. the transient receptor potential of vanilloid type-1 (TRPV1) channel, and, either directly or indirectly, cannabinoid receptors of type-1. Here we report the biochemical and pharmacological characterization of a so far neglected NAVAM, N-palmitoyl-vanillamide (palvanil), and propose its possible use instead of capsaicin, as a possible topical analgesic. Palvanil exhibited a kinetics of activation of human recombinant TRPV1-mediated intracellular calcium elevation significantly slower than that of capsaicin (t(1/2)=21s and 8s, respectively at 1μM). Slow kinetics of TRPV1 agonists were previously found to be associated with stronger potencies as TRPV1 desensitizing agents, which in turn are usually associated with lower pungency and stronger anti-hyperalgesic activity. Accordingly, palvanil desensitized the human recombinant TRPV1 to the effect of capsaicin (10nM) with significantly higher potency than capsaicin (IC(50)=0.8nM and 3.8nM, respectively), this effect reaching its maximum more rapidly (50 and 250min, respectively). Palvanil was also more potent than capsaicin at desensitizing the stimulatory effect of TRPV1 by low pH together with anandamide, which mimics conditions occurring during inflammation. In the eye-wiping assay carried out in mice, palvanil was not pungent and instead caused a strong and long-lasting inhibition of capsaicin-induced eye-wiping. Finally, intraplantar palvanil inhibited the second phase of the nociceptive response to formalin in mice. In conclusion, palvanil appears to be a non-pungent analogue of capsaicin with stronger desensitizing effects on TRPV1 and hence potentially higher anti-hyperalgesic activity. |
|
| In vivo: |
| Pharmacol Res . 2012 Sep;66(3):243-50 | | Palvanil, a non-pungent capsaicin analogue, inhibits inflammatory and neuropathic pain with little effects on bronchopulmonary function and body temperature[Pubmed: 22634607] | | N-Palmitoyl-vanillamide (palvanil) is a non-pungent capsaicinoid, found in low amounts in Capsicum and shown to rapidly desensitize transient receptor potential vanilloid type-1 (TRPV1) channels to the action of capsaicin and to exert analgesic effects after local administration. We have investigated here if systemic administration of palvanil to mice causes two typical adverse events of TRPV1 agonists, i.e. profound changes in body temperature and bronchoconstriction, and if it can still produce effective inhibition of inflammatory and chronic pain in different experimental models. Varying doses of palvanil were tested subcutaneously and acutely on body temperature in vivo or, or as a bolus, on bronchopulmunary function ex vivo, in comparison with capsaicin. Intraperitoneal palvanil was also tested against formalin-induced nocifensive behavior and carrageenan-induced oedema and thermal hyperalgesia, acutely, and against mechanical allodynia and thermal hyperalgesia in mice with spared nerve injury (SNI) of the sciatic nerve, after repeated administration over 7 days from SNI. Palvanil, at therapeutically relevant doses, produced significantly less hypothermia and bronchoconstriction than capsaicin. Palvanil (0.5-2.5 mg/kg) abolished formalin-induced nocifensive behavior and strongly attenuated SNI-induced mechanical allodynia and thermal hyperalgesia and carrageenan-induced oedema and thermal hyperalgesia. Systemic administration of the non-pungent capsaicinoid, palvanil, produces, at least in mice, much less of those side effects typical of TRPV1 agonists (hypothermia and bronchoconstriction), whilst being very effective at reducing pain and oedema. Thus, palvanil might be developed further as a novel pharmacological treatment for chronic abnormal pain. | | Pharmacol Res . 2012 Sep;66(3):243-250. | | Palvanil, a non-pungent capsaicin analogue, inhibits inflammatory and neuropathic pain with little effects on bronchopulmonary function and body temperature[Pubmed: 22634607] | | N-Palmitoyl-vanillamide (palvanil) is a non-pungent capsaicinoid, found in low amounts in Capsicum and shown to rapidly desensitize transient receptor potential vanilloid type-1 (TRPV1) channels to the action of capsaicin and to exert analgesic effects after local administration. We have investigated here if systemic administration of palvanil to mice causes two typical adverse events of TRPV1 agonists, i.e. profound changes in body temperature and bronchoconstriction, and if it can still produce effective inhibition of inflammatory and chronic pain in different experimental models. Varying doses of palvanil were tested subcutaneously and acutely on body temperature in vivo or, or as a bolus, on bronchopulmunary function ex vivo, in comparison with capsaicin. Intraperitoneal palvanil was also tested against formalin-induced nocifensive behavior and carrageenan-induced oedema and thermal hyperalgesia, acutely, and against mechanical allodynia and thermal hyperalgesia in mice with spared nerve injury (SNI) of the sciatic nerve, after repeated administration over 7 days from SNI. Palvanil, at therapeutically relevant doses, produced significantly less hypothermia and bronchoconstriction than capsaicin. Palvanil (0.5-2.5 mg/kg) abolished formalin-induced nocifensive behavior and strongly attenuated SNI-induced mechanical allodynia and thermal hyperalgesia and carrageenan-induced oedema and thermal hyperalgesia. Systemic administration of the non-pungent capsaicinoid, palvanil, produces, at least in mice, much less of those side effects typical of TRPV1 agonists (hypothermia and bronchoconstriction), whilst being very effective at reducing pain and oedema. Thus, palvanil might be developed further as a novel pharmacological treatment for chronic abnormal pain. | | Naunyn Schmiedebergs Arch Pharmacol . 2020 Aug;393(8):1357-1364. | | Desensitization of transient receptor potential vanilloid type-1 (TRPV1) channel as promising therapy of irritable bowel syndrome: characterization of the action of palvanil in the mouse gastrointestinal tract[Pubmed: 32002574] | | TRPV1 are involved in the control of the gastrointestinal (GI) functions and pain sensation. Their activation induces pain but it is followed by desensitization, which in turn causes analgesia. The studies from the last two decades indicate that TRPV1 are involved in visceral hypersensitivity in the GI tract and pathogenesis of irritable bowel syndrome (IBS). Therefore, the aim of this study is to assess the action of fast desensitizing agonist of TRPV1, palvanil (N-palmitoyl-vanillamine), in the murine GI tract and on nociception to evaluate its potential application in the therapy of IBS. The effect of palvanil on smooth muscle contractility was evaluated using organ baths. The impact of palvanil on intestinal secretion was assessed in Ussing chambers. In vivo, the action of palvanil (0.1-1 mg/kg) was assessed in whole GI transit, fecal pellet output, and colonic bead expulsion tests. The antinociceptive potency of palvanil was tested in the mustard oil-induced pain test. Palvanil inhibited colonic contractions (evoked by electrical field stimulation, EFS) and decreased the ion transport in the colon stimulated with forskolin. It did not affect secretion in experiments with veratridine. In vivo, palvanil prolonged whole GI transit at all doses tested. At the lower dose tested, it accelerated colonic motility during first 60 min following injection. By contrast, at the dose of 1 mg/kg, colonic motility was inhibited. Palvanil induced antinociceptive action at all tested doses in mustard oil-induced pain test. TRPV1 fast-desensitizing compounds, i.e., palvanil, may be promising agents in the therapy of IBS since it modulates intestinal motility and reduces visceral pain. | | Pharmacol Res . 2013 Oct;76:98-105. | | Piperazinyl carbamate fatty acid amide hydrolase inhibitors and transient receptor potential channel modulators as "dual-target" analgesics[Pubmed: 23911581] | | We showed previously that inhibiting fatty acid amide hydrolase (FAAH), an endocannabinoid degrading enzyme, and transient receptor potential vanilloid type-1 (TRPV1) channels with the same molecule, the naturally occurring N-arachidonoyl-serotonin (AA-5-HT), produces more efficacious anti-nociceptive and anti-hyperalgesic actions than the targeting of FAAH or TRPV1 alone. We also reported the synthesis of some piperazinyl carbamates as "dual" FAAH inhibitors and either antagonists at TRPV1 or agonists/desensitizers of the transient receptor potential ankyrin type-1 (TRPA1) cannel, another target for analgesic drugs. We investigated here if two such compounds, the FAAH/TRPV1 blocker OMDM198 and the FAAH inhibitor/TRPA1 agonist, OMDM202, exert anti-nociceptive actions in the formalin test of pain in mice, and through what mechanism. Both compounds inhibited the second phase of the response to formalin, the effect being maximal at 3 mg/kg, i.p. Antagonism of CB1 or CB2 receptors with AM251 or AM630 (1 mg/kg, i.p.), respectively, reversed this effect. A TRPV1 agonist, palvanil (0.1 mg/kg, i.p.), also reversed the analgesic effect of OMDM198. OMDM202 action was also antagonized by a per se inactive dose of the selective TRPA1 blocker, AP-18 (0.05 mg/kg, i.p.), but not by a TRPV1 antagonist. AP-18 at higher doses (0.1-0.2 mg/kg) inhibited both the first and second phase of the formalin response. The effects of OMDM198 and OMDM202 were accompanied by elevation of anandamide levels in the spinal cord. OMDM198 (0.1-5.0 mg/kg, i.p.) also reversed carrageenan-induced oedema and thermal hyperalgesia in mice with efficacy similar to that of AA-5-HT. These data suggest that "dual" fatty acid amide hydrolase and transient receptor potential channel modulators should be clinically evaluated as novel analgesics. |
|

 Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019)
Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019) Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019)
Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019) Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)
Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)







 ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019)
ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019) Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019)
Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019) Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)
Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}