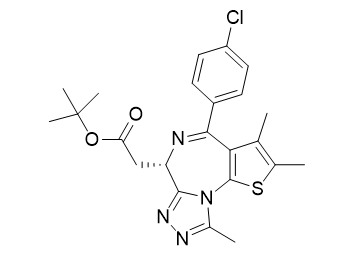
| Description: |
(+)-JQ1 is a BET bromodomain inhibitor, with IC50 of 77 nM/33 nM for BRD4(1/2) in cell-free assays, binding to all bromodomains of the BET family, but not to bromodomains outside the BET family. (+)-JQ1 suppresses cell proliferation via inducing autophagy. |
| Targets: |
Autophagy |
| In vitro: |
| Proc Natl Acad Sci U S A,2011 Oct 4;108(40):16669-74. | | Targeting MYC dependence in cancer by inhibiting BET bromodomains.[Pubmed: 21949397] | The MYC transcription factor is a master regulator of diverse cellular functions and has been long considered a compelling therapeutic target because of its role in a range of human malignancies. However, pharmacologic inhibition of MYC function has proven challenging because of both the diverse mechanisms driving its aberrant expression and the challenge of disrupting protein-DNA interactions.
METHODS AND RESULTS:
Here, we demonstrate the rapid and potent abrogation of MYC gene transcription by representative small molecule inhibitors of the BET family of chromatin adaptors. MYC transcriptional suppression was observed in the context of the natural, chromosomally translocated, and amplified gene locus. Inhibition of BET bromodomain-promoter interactions and subsequent reduction of MYC transcript and protein levels resulted in G(1) arrest and extensive apoptosis in a variety of leukemia and lymphoma cell lines. Exogenous expression of MYC from an artificial promoter that is resistant to BET regulation significantly protected cells from cell cycle arrest and growth suppression by BET inhibitors. MYC suppression was accompanied by deregulation of the MYC transcriptome, including potent reactivation of the p21 tumor suppressor. Treatment with a BET inhibitor resulted in significant antitumor activity in xenograft models of Burkitt's lymphoma and acute myeloid leukemia.
CONCLUSIONS:
These findings demonstrate that pharmacologic inhibition of MYC is achievable through targeting BET bromodomains. Such inhibitors may have clinical utility given the widespread pathogenetic role of MYC in cancer. |
|
| In vivo: |
| Cell,2011 Sep 16;146(6):904-17. | | BET bromodomain inhibition as a therapeutic strategy to target c-Myc.[Pubmed: 21889194] | MYC contributes to the pathogenesis of a majority of human cancers, yet strategies to modulate the function of the c-Myc oncoprotein do not exist.
METHODS AND RESULTS:
Toward this objective, we have targeted MYC transcription by interfering with chromatin-dependent signal transduction to RNA polymerase, specifically by inhibiting the acetyl-lysine recognition domains (bromodomains) of putative coactivator proteins implicated in transcriptional initiation and elongation. Using a selective small-molecule bromodomain inhibitor, JQ1, we identify BET bromodomain proteins as regulatory factors for c-Myc. BET inhibition by JQ1 downregulates MYC transcription, followed by genome-wide downregulation of Myc-dependent target genes.
CONCLUSIONS:
In experimental models of multiple myeloma, a Myc-dependent hematologic malignancy, JQ1 produces a potent antiproliferative effect associated with cell-cycle arrest and cellular senescence. Efficacy of JQ1 in three murine models of multiple myeloma establishes the therapeutic rationale for BET bromodomain inhibition in this disease and other malignancies characterized by pathologic activation of c-Myc. |
|

 Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019)
Cell. 2018 Jan 11;172(1-2):249-261.e12. doi: 10.1016/j.cell.2017.12.019.IF=36.216(2019) Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019)
Cell Metab. 2020 Mar 3;31(3):534-548.e5. doi: 10.1016/j.cmet.2020.01.002.IF=22.415(2019) Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)
Mol Cell. 2017 Nov 16;68(4):673-685.e6. doi: 10.1016/j.molcel.2017.10.022.IF=14.548(2019)







 ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019)
ACS Nano. 2018 Apr 24;12(4): 3385-3396. doi: 10.1021/acsnano.7b08969.IF=13.903(2019) Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019)
Nature Plants. 2016 Dec 22;3: 16206. doi: 10.1038/nplants.2016.205.IF=13.297(2019) Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019)
Sci Adv. 2018 Oct 24;4(10): eaat6994. doi: 10.1126/sciadv.aat6994.IF=12.804(2019){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}